


Seltene Erkrankungen
Ein Überblick & Unterstützung für Betroffene und Angehörige
Inhaltsverzeichnis
Was sind seltene Erkrankungen?
Seltene Erkrankungen sind Krankheiten, die weniger als 5 von 10.000 Menschen betreffen.¹ Trotz der individuellen Seltenheit gibt es insgesamt über 6.000 bekannte seltene Erkrankungen, von denen in Deutschland etwa 4 Millionen und weltweit 400 Millionen Menschen betroffen sind.¹,² Viele dieser Erkrankungen sind genetisch bedingt, schwer zu diagnostizieren und oft nur unzureichend erforscht. Dies führt zu langen Leidenswegen für Betroffene und ihre Angehörigen.
Die Diagnose seltener Erkrankungen stellt Mediziner*innen vor besondere Herausforderungen. Die Symptome treten häufig unspezifisch auf, was die richtige Diagnosestellung verzögert und den Zugang zu notwendigen Therapien erschwert.³ Aufgrund der geringen Fallzahlen fehlt es oft an ausreichenden Forschungsergebnissen und spezialisierten Behandlungsmöglichkeiten.
Ein weiteres Problem stellt die gesellschaftliche Wahrnehmung dar. Viele seltene Erkrankungen sind kaum bekannt, weshalb Betroffene und ihre Familien nicht nur mit der Krankheit selbst, sondern auch mit mangelndem Verständnis und sozialer Isolation zu kämpfen haben.³,⁴ Besonders betroffen sind auch pflegende Angehörige, die oft eine immense Belastung erleben.³
Seltene Lebererkrankungen – Eine unterschätzte Herausforderung
ChatGPT:
Auch seltene Lebererkrankungen stellen eine große Herausforderung für Patient*innen, Angehörige und das Gesundheitssystem dar. Insgesamt sind auch in Deutschland nicht wenige Menschen von diesen Erkrankungen betroffen, doch aufgrund ihrer Seltenheit werden sie oft erst spät diagnostiziert oder gar nicht erkannt.⁴,⁵ Die daraus resultierenden gesundheitlichen Folgen können gravierend sein, da eine frühzeitige Behandlung oft entscheidend für den Krankheitsverlauf ist.


Intensive Forschung für neue Therapieansätze
Ein zentrales Problem bei seltenen Lebererkrankungen ist der Mangel an gezielter Forschung und kausalen Therapien. Aufgrund der geringen Patientenzahlen ist es schwierig, entsprechende Studien zu initiieren und die Zulassung von Medikamenten zu erreichen. Auch die Finanzierung von Forschungsprojekten ist häufig begrenzt, und klinische Studien sind wegen der kleinen Fallzahlen besonders herausfordernd.³,⁶
Trotz dieser Hürden gibt es Hoffnung: Fortschritte in der Entwicklung innovativer Arzneimittel eröffnen neue Perspektiven für Betroffene. Genau hier setzt Ipsen an: Als biopharmazeutisches Unternehmen engagiert sich Ipsen gezielt für die Entwicklung neuer Therapieoptionen bei seltenen Lebererkrankungen. Durch internationale Forschungskooperationen, die Unterstützung von Patientenregistern und Investitionen in klinische Studien trägt Ipsen dazu bei, Wissen zu bündeln und neue Behandlungsansätze voranzutreiben.
Frühzeitige Diagnosen, spezialisierte Therapien und ein vertieftes Verständnis der Krankheitsmechanismen sind entscheidend, um die Lebensqualität der Betroffenen zu verbessern. Gleichzeitig setzt sich Ipsen dafür ein, dass Patient*innen und Angehörige Zugang zu fundierten Informationen, Netzwerken und Unterstützungsangeboten erhalten – für einen besseren Umgang mit der Erkrankung und mehr Lebensperspektive.
Seltene Lebererkrankungen im Fokus
Seltene Erkrankungen der Leber sind eine sehr heterogene Gruppe von Krankheitsbildern, die sich in ihrer Entstehung, ihren Ausprägungen und dem Verlauf zum Teil wesentlich unterscheiden. Einige beruhen auf genetischen Veränderungen oder anatomischen Fehlbildungen und zeigen sich bereits im Säuglings- oder Kindesalter. Andere treten erst im Erwachsenenleben auf – etwa im Rahmen autoimmuner Prozesse oder als Folge bestimmter Grunderkrankungen.⁷,⁸,⁹
Unterschiede gibt es auch hinsichtlich der betroffenen Leberfunktionen: So kann etwa die Entgiftungsfunktion der Leber gestört sein, der Stoffwechsel aus dem Gleichgewicht geraten oder die Syntheseleistung eingeschränkt sein. Bei einigen seltenen Krankheitsbildern liegt wiederum eine Störung im Galleabfluss vor. Man spricht daher auch von cholestatischen Lebererkrankungen.¹⁰ Drei dieser Erkrankungen werden in diesem Beitrag ausführlich vorgestellt.
Das Zusammenspiel von Leber und Galle
Um die seltenen cholestatischen Lebererkrankungen besser verstehen zu können, lohnt sich ein Blick auf die Funktionsweise von Leber und Galle. Die Leber als größtes inneres Organ ist quasi das Chemielabor des Körpers und übernimmt zahlreiche lebenswichtige Aufgaben. Alle über den Darm aufgenommenen Nährstoffe gelangen zunächst in die Leber und werden dort verstoffwechselt und teilweise gespeichert. Auch beim Energie- und Eiweißstoffwechsel spielt die Leber eine zentrale Rolle. Ebenso bei der Entgiftung, dem Abbau von Stoffwechselprodukten und Medikamenten.¹⁰,¹¹
Eine weitere Funktion der Leber ist die Produktion von Galle. Die gelblich-grüne Flüssigkeit enthält unter anderem Elektrolyte, Gallensalze, Cholesterin und andere Lipide sowie Abbauprodukte wie Bilirubin. Sie wird fortlaufend in den Leberzellen produziert und zunächst über ein feines Netz kleiner Gallengänge innerhalb der Leber gesammelt. Von dort gelangt sie in den Ductus hepaticus communis, den Hauptgallengang der Leber.
Die Galle wird dann über einen Ausführungsgang (Ductus choledochus) in den Zwölffingerdarm abgegeben, den obersten Abschnitt des Dünndarms. Sie kann alternativ in die Gallenblase geleitet werden, wo sie gespeichert und eingedickt wird – insbesondere zwischen den Mahlzeiten. Wird über die Nahrung ein Verdauungsreiz ausgelöst, zieht sich die Gallenblase zusammen und setzt die gespeicherte Galle frei.
Im Dünndarm übernimmt die Galle zwei zentrale Aufgaben: Zum einen sorgen die Gallensalze dafür, dass Fette aus der Nahrung in kleinere Bestandteile aufgespalten und besser aufgenommen werden können. Sie ist auch für die Aufnahme von fettlöslichen Vitaminen notwendig. Zum anderen werden über die Galle verschiedene Substanzen ausgeschieden, die der Körper nicht mehr benötigt, insbesondere überschüssiges Cholesterin und Bilirubin, ein Abbauprodukt des roten Blutfarbstoffs Hämoglobin.¹⁰,¹¹ Ein Teil der Gallensalze wird anschließend vom Darm wieder aufgenommen und von der Leber recycelt. Dies wird auch als enterohepatischer Kreislauf bezeichnet.
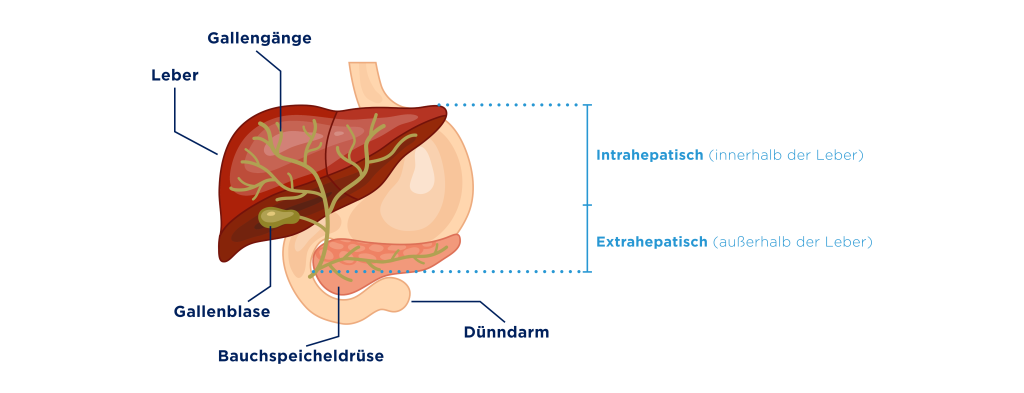
Damit das Zusammenspiel von Leber und Galle reibungslos funktioniert, müssen die Gallengänge durchgängig sein. Gerät der Gallefluss ins Stocken und kommt es zu einem Rückstau, kann das erhebliche gesundheitliche Auswirkungen nach sich ziehen.¹⁰
Seltene cholestatische Lebererkrankungen bei Kindern und Erwachsenen
Wenn der Abfluss der Galle gestört ist, spricht man in der Medizin von einer Cholestase. Das Wort kommt aus dem Griechischen und bedeutet sinngemäß „Gallenstau“. Je nachdem, wo die Cholestase entsteht, unterscheidet man zwischen einer intrahepatischen Cholestase, also einer Störung in den kleinen Gallenwegen der Leber, und einer extrahepatischen Cholestase, bei der der Gallefluss außerhalb der Leber – etwa in den größeren Gallengängen – behindert ist.¹⁰
Die seltenen Lebererkrankungen können auf verschiedene Ursachen zurückgehen, beispielsweise eine genetische Veränderung oder angeborene Fehlbildung der Gallenwege und daher bereits Neugeborene und Kleinkinder betreffen.
Progressive familiäre intrahepatische Cholestase (PFIC)
Progressive familiäre intrahepatische Cholestase (abgekürzt PFIC) ist der Oberbegriff für eine Gruppe sehr seltener, genetisch bedingter Lebererkrankungen, denen Störungen der Gallesekretion zugrunde liegen. Bekannt sind mittlerweile bis zu 13 verschiedene PFIC-Formen, die auf unterschiedliche Gendefekte zurückzuführen sind. Mit rund 70% tritt der PFIC Typ 2 am häufigsten auf.⁸
Der Name beschreibt wesentliche Merkmale dieser Krankheitsbilder:
- progressiv = fortschreitender Verlauf
- familiär = erblich bedingt
- intrahepatische Cholestase = Gallestau innerhalb der Leber bzw. in den dort verlaufenden Gallengängen
PFIC zeigt sich meistens schon im Säuglings- und frühen Kindesalter.¹² In manchen Fällen bleibt die Erkrankung bis zum Erwachsenenalter unerkannt. Man schätzt, dass nur eines von 50.000 bis 100.000 neugeborenen Kindern von PFIC betroffen ist.⁸ Die Erkrankung tritt bei Mädchen und Jungen gleichermaßen auf.¹²
Wie wird PFIC vererbt?
PFIC wird in der Regel autosomal-rezessiv vererbt.⁸ Das bedeutet: Beide Elternteile tragen eine genetische Veränderung in sich, ohne selbst erkrankt zu sein. Erst wenn ein Kind die veränderte Erbinformation von der Mutter und vom Vater erbt, kann es zur Erkrankung kommen. Die Wahrscheinlichkeit liegt dann bei 25 Prozent, das Risiko besteht bei jeder Schwangerschaft erneut.¹³
Frühe Symptome einer PFIC
Erste Anzeichen der Lebererkrankung sind¹²
ein ausgeprägter Juckreiz, der sich durch häufiges Scheuern und Kratzen äußert. Die Kinder sind unruhig, leicht reizbar und schlafen oft schlecht.
Gelbsucht (Ikterus): Der Farbstoff Bilirubin, der normalerweise mit der Galle ausgeschieden wird, reichert sich stattdessen im Körper an und färbt Haut und Schleimhäute gelblich.
Weitere Hinweise auf eine PFIC können z.B. ein heller, manchmal fettig glänzender Stuhl sowie eine verlangsamte körperliche Entwicklung sein. Kinderärzt*innen sprechen in solchen Fällen von Gedeihstörungen, also einem langsameren Wachstum und geringerer Gewichtszunahme als altersgerecht zu erwarten ist.¹⁴
Die genannten Symptome treten nicht bei allen erkrankten Kindern auf. Je nach PFIC-Typ und individueller Ausprägung kann das Beschwerdebild sehr unterschiedlich sein.⁸
Diagnose: Wie wird eine PFIC festgestellt?
Da PFIC sehr selten ist und sich anfangs eher durch unspezifische Symptome äußert, kann es bis zur endgültigen Diagnose einige Zeit dauern. Um PFIC sicher festzustellen und andere Lebererkrankungen auszuschließen, sind mehrere Untersuchungsschritte erforderlich:⁸,¹⁵
Bluttest: Typische Leberwerte wie γ-Glutamyltransferase, Gallensäuren, Bilirubin, Transaminasen, Albumin und Gerinnungsfaktoren werden überprüft.
Ultraschalluntersuchung: Mittels Sonografie lassen sich Lebergröße, Struktur und Durchblutung erfassen sowie Veränderungen an Gallenwegen und Milz erkennen.
Leberbiopsie: Unter Dämmerschlaf und Sedierung wird eine kleine Gewebeprobe entnommen. Diese gibt Hinweise auf das Ausmaß der Leberschädigung und hilft, andere Ursachen der Erkrankung auszuschließen.
Gentest: Dient zur Bestätigung der Diagnose und Bestimmung des PFIC-Subtyps. Dabei wird nach krankheitsauslösenden Veränderungen im Erbgut gesucht. Oft werden auch Blutproben der Eltern mit einbezogen.
Die Abklärung erfolgt idealerweise in einem spezialisierten Zentrum, das über Erfahrung mit seltenen Lebererkrankungen verfügt. Adressen und Kontaktdaten entsprechender Einrichtungen in Wohnortnähe sind z.B. auf dem Portal Deutsche Leberhilfe e.V. zu finden.
Therapieoptionen bei PFIC
Die Behandlung zielt darauf ab, den gestörten Gallefluss zu verbessern, Beschwerden zu lindern und Folgeschäden der Leber vorzubeugen. Welche Maßnahmen zur Anwendung kommen, hängt vom PFIC-Typ, der individuellen Symptomatik und dem Krankheitsverlauf ab.⁸
- Zur medikamentösen Behandlung bei PFIC stehen Hemmstoffe der Wiederaufnahme von Gallensäuren aus dem Darm, sogenannte IBAT-Inhibitoren, zur Verfügung. Sie setzen im letzten Abschnitt des Dünndarms an und unterbrechen den Rücktransport der Gallensäuren zur Leber. Dadurch wird die Leber entlastet, und der Gallensäurespiegel im Blut kann sinken – was insbesondere den Juckreiz mindern kann. MEHR ERFAHREN
- Besonderes Augenmerk gilt der Ernährung. Da die Aufnahme der fettlöslichen Vitamine (A, D, E und K) beeinträchtigt ist, ist eine gezielte Substitution notwendig. Auch der Energiebedarf ist erhöht, gleichzeitig verspüren Kinder mit PFIC oft weniger Appetit. Eine Ernährungstherapie kann helfen, Mangelzustände zu vermeiden und eine altersgerechte Entwicklung zu unterstützen.
- Akute Linderung bei starkem Juckreiz können zusätzlich feuchtigkeitsspendende, rückfettende Salben verschaffen.
Reichen Medikamente nicht aus, um den gestörten Gallefluss ausreichend zu verbessern und Beschwerden zu lindern, kann ein chirurgischer Eingriff erwogen werden. Ziel ist es, die Wiederaufnahme der Gallensäuren im Darm zu verringern und die Leber langfristig zu entlasten. Ob eine solche Maßnahme infrage kommt, hängt von Verlauf, PFIC-Typ und individueller Krankengeschichte ab.⁸
Unabhängig von der Therapie kann es im Krankheitsverlauf zu einer fortschreitenden Schädigung der Leber kommen und eine Lebertransplantation notwendig werden. Ausführliche Informationen zur Transplantation im Kindesalter stellt die Patientenorganisation Leberkrankes Kind e.V. bereit.
Gallengangatresie bei Säuglingen
Die Gallengangatresie ist eine seltene Lebererkrankung, die bereits bei Neugeborenen auftritt.⁷ In der medizinischen Fachsprache wird die Erkrankung auch als biliäre Atresie bezeichnet – ein Begriff, der sich auf einen Verschluss der Gallenwege innerhalb oder außerhalb der Leber bezieht. Dabei ist der Abfluss der Galle aus der Leber in den Darm erheblich gestört – mit schwerwiegenden gesundheitlichen Folgen. Umso wichtiger sind eine frühzeitige Diagnose und umgehende Behandlung.
Schätzungen zufolge tritt die Erkrankung in Deutschland bei einem von 15.000 Neugeborenen auf.¹⁴
Ursachen und Entstehung
Warum es bei manchen Säuglingen zu einer Gallengangatresie kommt, ist bislang nicht vollständig geklärt. Die Gallenwege können entweder schon vor der Geburt nicht richtig angelegt worden sein oder sie vernarben kurz danach aus noch unbekannten Gründen.
Fachleute vermuten, dass mehrere Faktoren zusammenspielen. In einigen Fällen scheinen Infektionen während der Schwangerschaft oder kurz nach der Geburt eine Rolle zu spielen, möglicherweise ausgelöst durch bestimmte Viren. Auch eine Fehlsteuerung des Immunsystems wird diskutiert. Eine eindeutig erbliche Ursache ist eher selten.¹⁶
Symptome einer Gallengangatresie
Neugeborene mit biliärer Atresie erscheinen in den ersten Lebenstagen zunächst gesund und entwickeln sich altersgerecht.⁷ Selbst eine leichte Gelbfärbung von Haut und Augen – auch Neugeborenen-Gelbsucht oder Neugeborenen-Ikterus genannt – ist in den ersten zehn bis 14 Tagen nach der Geburt ein häufiges und in der Regel harmloses Phänomen.⁷
Ein erstes Warnzeichen kann sein, wenn die Gelbsucht nach zwei Wochen weiterhin besteht. Auch die Beschaffenheit der Ausscheidungen kann Hinweise auf eine Cholestase geben. Ein typisches Symptom ist ein ungewöhnlich heller oder farbloser (acholischer) Stuhl, da der Gallenfarbstoff Bilirubin fehlt, der ihn normalerweise bräunlich färbt.¹⁴ Der Urin kann dagegen deutlich dunkler aussehen als üblich, was darauf hindeutet, dass Bilirubin vermehrt über die Nieren ausgeschieden wird statt mit der Galle in den Dünndarm zu gelangen.
Wie wird eine Gallengangatresie festgestellt?
Bei anhaltender Gelbsucht und/oder auffälliger Farbe von Stuhl und Urin wird der Kinderarzt / die Kinderärztin das Gesamtbilirubin und den Anteil des konjugierten Bilirubins im Blut bestimmen. Ist dieser Wert erhöht, sollte umgehend eine weiterführende Diagnostik in einer Facharztpraxis für Kindergastroenterologie oder einem spezialisierten Zentrum für Lebererkrankungen im Kindesalter erfolgen.⁷
Um eine Gallengangatresie zu bestätigen oder auszuschließen, werden weitere Bluttests, eine Ultraschalluntersuchung und eine Leberbiopsie vorgenommen. Bei einem starken Verdacht auf eine Gallengangatresie wird zur Bestätigung der Diagnose ein kleiner operativer Eingriff durchgeführt. Bei der Intraoperativen Cholangiographie wird ein Kontrastmittel in die Gallengänge eingebracht, um den Gallefluss sichtbar zu machen. Falls sich der Verdacht bestätigt, kann in demselben Eingriff bereits die sogenannte Kasai-Operation erfolgen.¹⁷,¹⁸
Behandlung der Gallengangatresie
Die Gallengangatresie ist nicht heilbar. Ziel der Behandlung ist es, den gestörten Galleabfluss aus der Leber wiederherzustellen und ein Fortschreiten der Erkrankung zu verhindern oder zumindest zu verzögern. Eine kausale medikamentöse Therapie gibt es bislang noch nicht, einzige Behandlungsmöglichkeit ist die Kasai-Operation, die jedoch nur bei extrahepatischem Verschluss angewandt werden kann. Dabei werden die undurchlässigen Gallengänge entfernt und ein künstlicher Weg geschaffen, über den die Galle direkt in den Dünndarm abfließen kann. Der Eingriff sollte möglichst früh erfolgen – die größten Erfolgsaussichten bestehen bei einer Operation in den ersten 30 bis 60 Lebenstagen des Kindes.⁷,¹⁶
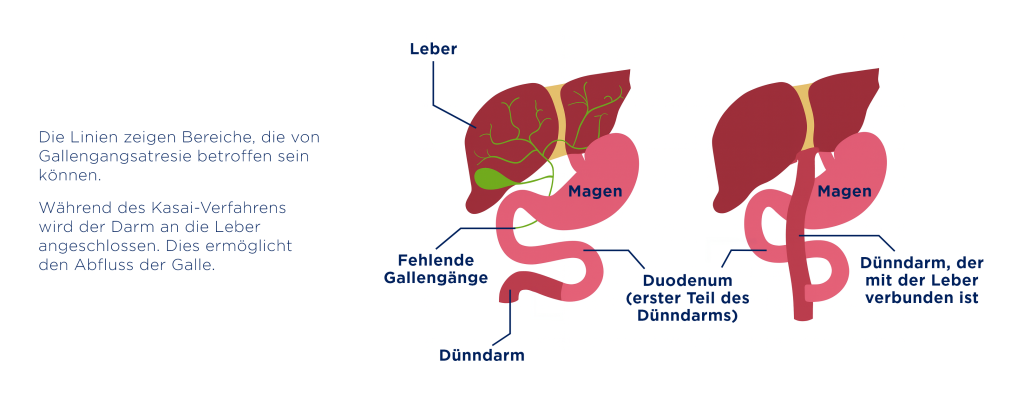
Nach der Operation ist weiterhin eine engmaschige medizinische Betreuung notwendig, um mögliche Komplikationen frühzeitig zu erkennen und zu behandeln. Dazu gehören regelmäßige Kontrollen der Leberwerte, bildgebende Untersuchungen und ggfs. die Gabe entzündungshemmender oder antibiotischer Medikamente. Schreitet die Erkrankung weiter fort und entwickelt sich eine Zirrhose, kann eine Lebertransplantation erforderlich werden.⁷
Leben mit PFIC und Gallengangatresie
Den seltenen Lebererkrankungen bei Kindern liegen zwar verschiedene Ursachen zugrunde, die Symptome und Herausforderungen ähneln sich jedoch häufig. Welche Maßnahmen lindern den anhaltenden Juckreiz? Was ist bei möglichen Wachstums- und Gedeihstörungen hinsichtlich der Ernährung zu beachten? Wie bereitet man sich auf den Arztbesuch vor? Die Broschüre „Leben mit seltenen Erkrankungen“ des Vereins Leberkrankes Kind e.V. und der Gesellschaft für Pädiatrische Gastroenterologie und Ernährung e.V. (GPGE) gibt praktische Empfehlungen zum Symptommanagement bei PFIC und Gallengangatresie und greift Themen auf, die viele Familien im Alltag beschäftigen – etwa den Umgang mit den emotionalen Belastungen einer chronischen Erkrankung sowie Fragen zu sozialrechtlichen Ansprüchen und finanzieller Unterstützung.
Primär biliäre Cholangitis (PBC): Eine seltene Lebererkrankung bei Erwachsenen
Die primär biliäre Cholangitis (kurz PBC) gilt als die häufigste Form der seltenen cholestatischen Lebererkrankungen bei Erwachsenen. Sie tritt überwiegend bei Frauen zwischen 40 und 60 Jahren (90 %) auf. Es können aber auch Männer oder Erwachsene jüngeren Alters betroffen sein.⁹ Bei Kindern wird PBC nicht beobachtet.
Die primär biliäre Cholangitis zählt zu den Autoimmunerkrankungen.⁹ Das bedeutet, dass sich das körpereigene Abwehrsystem nicht nur gegen Eindringlinge wie Viren und Bakterien richtet, sondern fälschlicherweise auch die eigenen gesunden Körperzellen angreift. Im Fall der PBC sind das die kleinen Gallengänge innerhalb der Leber, die durch eine chronische Entzündung nach und nach geschädigt werden.⁹
Die PBC ist eine chronisch fortschreitende Erkrankung, die jedoch individuell ganz unterschiedlich verlaufen kann.
- Typische, jedoch für PBC nicht spezifische Symptome sind anhaltende Müdigkeit (Fatigue) oder Juckreiz. Im frühen Stadium treten häufig noch keine Symptome auf.⁹,¹⁹,²⁰
- In späteren Stadien kann es zu einer Fibrose – einer krankhaften Bildung von Bindegewebe in der Leber – und in manchen Fällen zu einer Leberzirrhose kommen.⁹,²¹
Die Diagnose basiert meist auf einer Kombination aus typischen Laborwerten, dem Nachweis spezifischer autoimmuner Antikörper sowie bildgebenden Verfahren. In Einzelfällen kann eine Leberbiopsie notwendig sein, um den Entzündungsgrad und das Ausmaß der Gewebeschädigung zu beurteilen.⁹,²¹
PBC ist nicht heilbar, es stehen aber medikamentöse Behandlungsoptionen zur Verfügung, die den Krankheitsverlauf verlangsamen oder sogar aufhalten können. Dazu gehören insbesondere Präparate, die die Zusammensetzung der Gallensäuren und/oder den Entzündungsprozess günstig beeinflussen. In manchen Fällen kommen noch zusätzliche Therapien zum Einsatz – etwa bei unzureichendem Ansprechen auf die Standardbehandlung oder wenn Symptome wie z.B. Juckreiz als sehr belastend empfunden werden.⁹,²¹
Eine seltene Erkrankung wie die PBC ruft viele Fragen auf. Weiterführende Informationen zu Diagnoseverfahren, Therapieoptionen, einen leichteren Umgang mit den Symptomen im Alltag und Strategien zur Erhaltung der Lebensqualität haben wir in unserem Beitrag „Primär biliäre Cholangitis“ zusammengestellt.
Vielfältige Herausforderungen für Betroffene und Angehörige
Menschen mit seltenen Lebererkrankungen sind nicht nur mit gesundheitlichen Problemen konfrontiert – oft belastet die gesellschaftliche Wahrnehmung noch zusätzlich. Ein weitverbreitetes Vorurteil ist die Annahme, Lebererkrankungen seien grundsätzlich selbstverschuldet, etwa durch Alkoholmissbrauch oder einen ungesunden Lebensstil.⁶ Diese Fehleinschätzung führt zu einer Stigmatisierung, die viele Betroffene tief verletzt – insbesondere dann, wenn die Erkrankung genetisch bedingt ist oder auf andere, nicht beeinflussbare Ursachen zurückgeht.
Viele Erkrankte berichten davon, dass sie sich immer wieder rechtfertigen müssen, selbst in Gesprächen mit medizinischem Personal oder im sozialen Umfeld. Das Gefühl, für die eigene Krankheit verantwortlich gemacht zu werden, führt häufig zu Scham, sozialem Rückzug und psychischen Problemen.
Auch Angehörige tragen eine enorme körperliche und emotionale Last, da sie oft über Jahre hinweg für ein krankes Familienmitglied sorgen. Die Herausforderungen sind vielfältig: Von der Organisation medizinischer Betreuung über den psychischen Druck bis hin zur gesellschaftlichen Stigmatisierung, die mit Lebererkrankungen einhergeht. Häufig müssen Angehörige ihren gesamten Alltag neu strukturieren, ihre berufliche Tätigkeit einschränken oder aufgeben und finanzielle Einbußen hinnehmen.²²
Ein weiteres Problem ist das geringe öffentliche Wissen über seltene Lebererkrankungen. Die Unsichtbarkeit der Erkrankung und der fehlende Austausch mit anderen Betroffenen verstärken das Gefühl der Isolation. Dabei wäre ein offener, aufgeklärter Umgang mit der Thematik entscheidend, um mehr Akzeptanz, Verständnis und gesellschaftliche Unterstützung zu schaffen.
Organisationen wie die Deutsche Leberhilfe, die Deutsche Leberstiftung, der Verein Leberkrankes Kind oder Kautz⁵ leisten hier bereits wertvolle Aufklärungsarbeit und sind wichtige Anlaufstellen für Betroffene und ihre Familien.
Auch die Initiative „Räume zum Reden“ setzt sich aktiv für Menschen mit seltenen Lebererkrankungen und ihre Angehörigen ein. Ziel ist es, das Bewusstsein für diese Erkrankungen zu stärken, Vorurteile abzubauen und ein Umfeld zu etablieren, in dem niemand mit seinen Sorgen allein bleibt. Die Plattform bietet pflegenden Angehörigen und Betroffenen fundierte Informationen, Austauschmöglichkeiten und praktische Unterstützung im Alltag. Durch die Zusammenarbeit mit Patientenorganisationen, Fachleuten und Pflegekräften fördert die Initiative Dialog, vernetzt wichtige Akteure und trägt dazu bei, Versorgungslücken zu schließen.
DAS KÖNNTE SIE INTERESSIEREN

Angehörigen-Report Seltene Erkrankungen
Welche Bedürfnisse und Wünsche haben Angehörige, die Menschen mit seltenen Erkrankungen unterstützen?



Primär Biliäre Cholangitis (PBC)
Mit aktivem Symptom-Management für eine bessere Lebensqualität.

PBC-Patienten Isabella im Interview
Patienten Isabella im Interview: 2021 erhielt sie die Diagnose, dass sie an primär biliärer Cholangitis (PBC) erkrankt ist.
Glossar
Acholisch
Fehlende oder deutlich verminderte Gallenfarbstoffe im Stuhl, der dadurch hell erscheint.
Albumin
Wichtiges Eiweiß im Blut, das in der Leber gebildet wird und unter anderem für den Transport von Stoffen und die Aufrechterhaltung des Blutvolumens sorgt.
Autoimmunerkrankung
Eine Erkrankung, bei der das Immunsystem irrtümlich körpereigene Zellen angreift.
Autosomal-rezessiv
Erbgang, bei dem eine Erkrankung nur auftritt, wenn beide Elternteile ein verändertes Gen vererben.
Biliär
Die Gallenwege betreffend.
Bilirubin
Abbauprodukt des roten Blutfarbstoffs Hämoglobin, das über die Galle ausgeschieden wird. Erhöhte Werte können zu Gelbsucht (Ikterus) führen.
Cholangitis
Entzündung der Gallengänge.
Cholestase
Störung des Galleflusses, bei der sich die Galle in verschiedenen Abschnitten der Gallenwege stauen kann.
Cholestatische Lebererkrankungen
Erkrankungen, bei denen der gestörte Abfluss der Galle eine zentrale Rolle spielt – entweder innerhalb (intrahepatisch) oder außerhalb (extrahepatisch) der Leber.
Cholesterin
Fettähnlicher Stoff, der im Körper für den Aufbau von Zellen und die Produktion von Gallensäuren und Hormonen gebraucht wird. Ein Teil des Cholesterins wird über die Nahrung aufgenommen, der größte Teil vom Körper selbst hergestellt.
Ductus choledochus
Hauptgallengang, der die Galle aus Leber und Gallenblase zum Dünndarm leitet.
Ductus hepaticus communis
Gallengang, der die in der Leber gebildete Galle sammelt und weiterleitet.
Eiweißsynthese
Bildung lebenswichtiger Eiweiße durch die Leber.
Enterohepatischer Kreislauf
Natürlicher Kreislauf, bei dem Gallensäuren nach der Fettverdauung im Darm wieder in die Leber zurückgeführt und dort erneut für die Gallenbildung verwendet werden.
Extrahepatische Cholestase
Gallenstau, dessen Ursache außerhalb der Leber liegt, meist in den größeren Gallengängen.
Fatigue
Anhaltende Müdigkeit oder Erschöpfung, die sich durch Ruhe oder Schlaf nicht ausreichend bessert.
Fibrose
Vermehrte Bildung von Bindegewebe in der Leber als Folge anhaltender Schädigung.
Gallensalze
Bestandteile der Galle, die bei der Fettverdauung eine wichtige Rolle spielen.
Gallesekretion
Bildung und Abgabe der Galle durch die Leber.
Gerinnungsfaktoren
Eiweiße, die für die Blutgerinnung wichtig sind und in der Leber gebildet werden.
Hämoglobin
Blutfarbstoff in den roten Blutkörperchen, der Sauerstoff transportiert.
IBAT-Inhibitoren
Medikamente, die die Wiederaufnahme von Gallensäuren im Darm blockieren (IBAT = Ileal Bile Acid Transporter). Dadurch verringert sich der Rückfluss von Gallensäuren zur Leber, was den Juckreiz bei cholestatischen Lebererkrankungen lindern kann.
Ikterus
Gelbfärbung von Haut, Schleimhäuten und Augen durch erhöhte Bilirubinwerte im Blut.
Intrahepatische Cholestase
Gallenstau, dessen Ursache innerhalb der Leber liegt.
Intraoperative Cholangiographie
Röntgendarstellung der Gallenwege während einer Operation.
Kasai-Operation
Die von dem japanischen Kinderchirurgen Morio Kasai entwickelte Operationstechnik wird bei Neugeborenen mit fehlenden oder verschlossenen Gallengängen (Gallengangsatresie) angewendet. Bei der Kasai-Operation (Hepatoporto-Enterostomie) wird eine direkte Verbindung zwischen Leber und Dünndarm hergestellt, um einen Abfluss der Galle zu ermöglichen.
Konjugiertes Bilirubin
Form von Bilirubin, das in der Leber wasserlöslich gemacht wurde, um mit der Galle ausgeschieden zu werden.
Leberbiopsie
Gewebeentnahme aus der Leber zur genaueren Untersuchung.
Lebertransplantation
Chirurgischer Eingriff, bei dem eine schwer geschädigte und nicht mehr ausreichend funktionsfähige Leber durch eine gesunde Spenderleber ersetzt wird.
Leberzirrhose
Spätfolge chronischer Lebererkrankungen, bei der die Leber zunehmend vernarbt und ihre Funktion stark eingeschränkt ist.
Lipide
Fette und fettähnliche Substanzen im Körper.
Neugeborenen-Gelbsucht
Häufige, meist harmlose Gelbfärbung der Haut und Augen bei Neugeborenen. Sie entsteht in den ersten Lebenstagen, da einerseits viele rote Blutkörperchen abgebaut werden, andererseits die Leber aber noch nicht die Kapazität hat, das dabei entstehende Bilirubin vollständig zu verarbeiten.
Pruritus
Medizinischer Fachbegriff für Juckreiz.
Sonografie
Ultraschalluntersuchung.
Transaminasen
Leberenzyme (z. B. ALT und AST), die bei Schädigung von Leberzellen ins Blut freigesetzt werden und als Leberwerte im Bluttest bestimmt werden.
Ursodeoxycholsäure (UDCA)
Ein Arzneistoff, der zur Behandlung cholestatischer Lebererkrankungen wie der PBC eingesetzt wird. UDCA ist eine Gallensäure, die für medizinische Zwecke synthetisch hergestellt wird. Sie kommt in sehr geringen Mengen auch natürlicherweise im menschlichen Körper vor. UDCA verbessert den Gallefluss, verdrängt schädliche Gallensäuren und wirkt entzündungshemmend auf die Gallengänge.
Quellen:
- Bundesministerium für Gesundheit. o. J. „Seltene Erkrankungen“. https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html. (Aufgerufen am 28.04.2025)
- Boycott, Kym M, und Roberto Giugliani. 2025. „The RDI–Lancet Commission on Rare Diseases: Improving Visibility to Address Health-Care Disparities for 400 Million People“. The Lancet 405 (10479): 605–7. https://doi.org/10.1016/S0140-6736(25)00211-9.
- Eidt, Daniela, Martin Frank, et. al. 2009. „Maßnahmen zur Verbesserung der gesundheitlichen Situation von Menschen mit Seltenen Erkrankungen in Deutschland“. Hannover: Leibniz Universität Hannover, Forschungsstelle für Gesundheitsökonomie. https://www.bundesgesundheitsministerium.de/fileadmin/Dateien/5_Publikationen/Praevention/Berichte/110516_Forschungsbericht_Seltene_Krankheiten.pdf. (Aufgerufen am 28.04.2025)
- Aichinger, Heike, Nicole Brkic, Diana Schneider, und Tanja Bratan. 2023. „Die gesundheitliche Situation von Menschen mit Seltenen Erkrankungen in Deutschland. Endbericht“. https://publica.fraunhofer.de/handle/publica/448247. (Aufgerufen am 28.04.2025)
- Deutsche Leberstiftung. 2025. „Tag der Seltenen Erkrankungen: Deutsche Leberstiftung stellt Herausforderungen seltener Lebererkrankungen in den Fokus“. https://www.deutsche-leberstiftung.de/site/assets/files/5521/dls_presse_pm_25-05_tag_seltene_erkrankungen_final_2025-02-21.pdf. (Aufgerufen am 29.04.2025)
- Jones, David E.J., Ekkehard Sturm, und Ansgar W. Lohse. 2018. „Access to Care in Rare Liver Diseases: New Challenges and New Opportunities“. Journal of Hepatology 68 (3): 577–85. https://doi.org/10.1016/j.jhep.2017.11.004
- Grothues, Dirk, Harald Engelhardt, Orsolya Genzel-Boroviczeny, Marion Gnädig, Milena Harm, André Hörning, Oliver Muensterer, et al. 2020. „S2k Leitlinie Cholestase im Neugeborenenalter, AWMF-Register Nr. 068/015“. https://register.awmf.org/assets/guidelines/068-015l_S2k_Cholestase-im-Neugeborenenalter_2022-08.pdf (Aufgerufen am 28.06.2025)
- Hudert, Christian A, Martin Jankofsky, Verena Keitel-Anselmino, Marcin Krawczyk, Andreas Kremer, Frank Lammert, Eberhard Lurz, et al. 2025. „S3-Leitlinie ‚Seltene Lebererkrankungen (LeiSe LebEr) – Genetisch-cholestatische Lebererkrankungen‘ der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS): April 2025 – AWMF-Registernummer: 021 – 027“. https://register.awmf.org/assets/guidelines/021-027l_S3_KF_Seltene-Lebererkrankungen-LeiSeLebEr-Lebererkrankungen-Paediatrie-bis-Erwachsenenalter_2025-05.pdf
- Sebode, Marcial, Heike Bantel, et. al. 2025. „S3-Leitlinie ‚Seltene Lebererkrankungen (LeiSe LebEr) – Autoimmune Lebererkrankungen von der Pädiatrie bis zum Erwachsenenalter‘ der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS): Februar 2025 – AWMF-Registernummer: 021–027“. Zeitschrift für Gastroenterologie 63 (06): 604–88. https://doi.org/10.1055/a-2558-5204.
- Manns, Michael P., und Sabine Schneidewind, Hrsg. 2016. Praxis der Hepatologie. Berlin, Heidelberg: Springer Berlin Heidelberg. https://doi.org/10.1007/978-3-642-41620-0.
- Schmidt, Robert F., Gerhard Thews, und Florian Lang, Hrsg. 2000. Physiologie des Menschen. Springer-Lehrbuch. Berlin, Heidelberg: Springer Berlin Heidelberg. https://doi.org/10.1007/978-3-662-09346-7.
- Srivastava, Anshu. 2014. „Progressive Familial Intrahepatic Cholestasis“. Journal of Clinical and Experimental Hepatology 4 (1): 25–36. https://doi.org/10.1016/j.jceh.2013.10.005.
- Arnemann, J. 2018. „Autosomal-rezessive Vererbung“. In Lexikon der Medizinischen Laboratoriumsdiagnostik, herausgegeben von Axel M. Gressner und Torsten Arndt, 1–1. Springer Berlin Heidelberg. https://doi.org/10.1007/978-3-662-49054-9_3442-1.
- Lurz, Eberhard, und Philip Bufler. 2021. „Neonatale Cholestase: Hintergrund, Diagnostik und Therapie“. Monatsschrift Kinderheilkunde 169 (3): 275–89. https://doi.org/10.1007/s00112-020-01042-3
- McKiernan, Patrick, Jesus Quintero Bernabeu, Muriel Girard, Giuseppe Indolfi, Eberhard Lurz, und Palak Trivedi. 2024. „Opinion Paper on the Diagnosis and Treatment of Progressive Familial Intrahepatic Cholestasis“. JHEP Reports 6 (1): 100949. https://doi.org/10.1016/j.jhepr.2023.100949.
- Antala, Swati, und Sarah A. Taylor. 2022. „Biliary Atresia in Children“. Clinics in Liver Disease 26 (3):341–54. https://doi.org/10.1016/j.cld.2022.03.001.
- Madadi-Sanjani, Omid, Bianca Hegen, Jun Oh, Konrad Reinshagen, und Christian Tomuschat. 2024. „Neues zur adjuvanten Therapie der Gallengangsatresie nach Kasai-Hepatoportoenterostomie“. Monatsschrift Kinderheilkunde, Dezember. https://doi.org/10.1007/s00112-024-02093-6.
- Madadi-Sanjani, Omid, Uta Herden, und Marie Uecker. 2025. „Die Kasai-Hepatoportoenterostomie zur Behandlung der Gallengangatresie – Worauf kommt es an?“ Die Chirurgie 96 (6): 474–81. https://doi.org/10.1007/s00104-025-02259-2.
- Brand, M., und A. E. Kremer. 2022. „Systemischer Pruritus: Was gibt es Neues in Diagnostik und Therapie?“ Die Dermatologie 73 (8): 600–608. https://doi.org/10.1007/s00105-022-05027-z.
- Faisal, Asma. 2024. „Understanding Fatigue and Pruritus in Primary Biliary Cholangitis“. Clinical Liver Disease 23 (1). https://doi.org/10.1097/CLD.0000000000000216.
- Steinmann, Silja, und Christoph Schramm. 2024. „Primär biliäre Cholangitis – Response-Kriterien der Erstlinientherapie und Perspektiven der Zweitlinientherapie“. Die Innere Medizin 65 (4): 340–46.https://doi.org/10.1007/s00108-024-01674-7.
- Mighiu, Claudia, Sonia O’Hara, Enrico Ferri Grazzi, Karen F. Murray, Jörn M. Schattenberg, Emily Ventura, Melanie Karakaidos, et al. 2022. „Impact of Progressive Familial Intrahepatic Cholestasis on Caregivers: Caregiver-Reported Outcomes from the Multinational PICTURE Study“. Orphanet Journal of Rare Diseases 17 (1): 32. https://doi.org/10.1186/s13023-022-02177-0